




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
In the nucleus of eukaryotic cells, the three-dimensional structure of chromosomes is akin to a sophisticated information network, with histone modification serving as the most active regulatory node within this network. Over the past decade, researchers have come to recognize that the dynamic modification of histones functions not only as an "on/off" switch for gene expression, but also as a critical link between epigenetic regulation and disease treatment. The spatial and temporal dynamics of these chemical modifications, ranging from acetylation to methylation to phosphorylation, are crucial in determining cell fate. Emerging technologies hold promise in overcoming the limitations of traditional research methods. However, it is essential to address the opportunities and challenges associated with clinical translation. In this article, we will systematically examine the dynamic characteristics of histone modifications, spanning from molecular mechanisms to clinical applications.
The chemical diversity of histone modifications has been shown to constitute the "grammar library" of epigenetic regulation, with its dynamic balance directly determining chromatin accessibility and gene expression patterns. In recent years, with the advent of ultra-high resolution microscopic imaging technology, researchers have found that modifications at the same locus may exhibit distinct functions due to cell cycle stages or microenvironmental signals. This spatio-temporal specificity provides a molecular basis for precise regulation.
H3K27ac: a spatiotemporal code for enhancer activation
H3K27 acetylation (H3K27ac) is distinctive due to its robust correlation with enhancer activity. In embryonic stem cells, CRISPR interference experiments demonstrated that knockdown of H3K27 acetyltransferase P300 results in the inactivation of approximately 70% of enhancers, while the level of H3K27ac in the promoter region decreases by only 15%. This selective dependence suggests that H3K27ac may promote the co-cohesion of transcription factors (e.g., OCT4) with the mediator complex (Mediator) through the formation of a "liquid-liquid phase-separated" microenvironment. In the context of breast cancer cells, for instance, abnormally elevated levels of H3K27ac in oncogenic enhancers drive sustained expression of MYC genes by recruiting BET proteins. This process can be specifically blocked by JQ1 inhibitors.
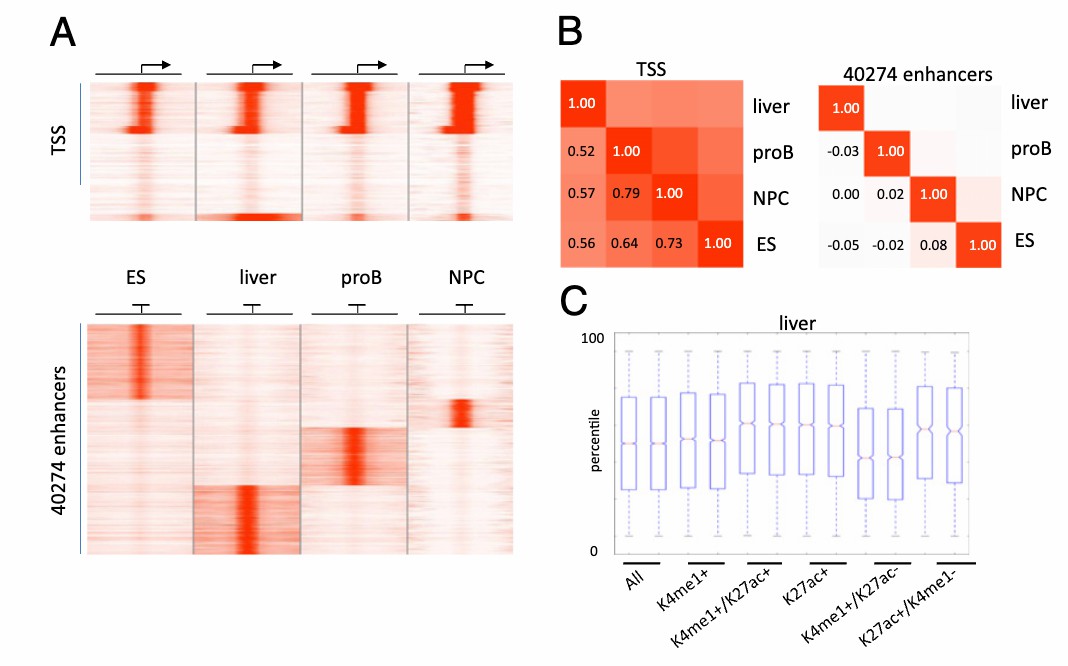 Enhanced proximal gene activity of distalH3K4me1-enriched regions is a function of H3K27ac (Creyghton et al., 2010)
Enhanced proximal gene activity of distalH3K4me1-enriched regions is a function of H3K27ac (Creyghton et al., 2010)
H4K16ac: an epigenetic clock in the aging process
The three major hallmarks of cellular senescence are the deletion of H4K16 acetylation (H4K16ac), telomere shortening, and nuclear fibrillar laminin abnormalities. Mechanistic studies have revealed that SIRT6 deacetylase regulates telomere heterochromatin formation by targeting H4K16ac. In SIRT6 knockout mice, elevated levels of H4K16ac lead to chromatin loosening in the telomere region, which triggers telomere dysfunctional fusion (TDF) and ultimately accelerates the senescence phenotype. Notably, the administration of an AAV vector to deliver the SIRT6 gene has been shown to restore H4K16ac homeostasis in liver cells of aged mice, thereby extending their healthy lifespan by up to 30%. This finding provides compelling evidence for the potential of anti-aging therapies.
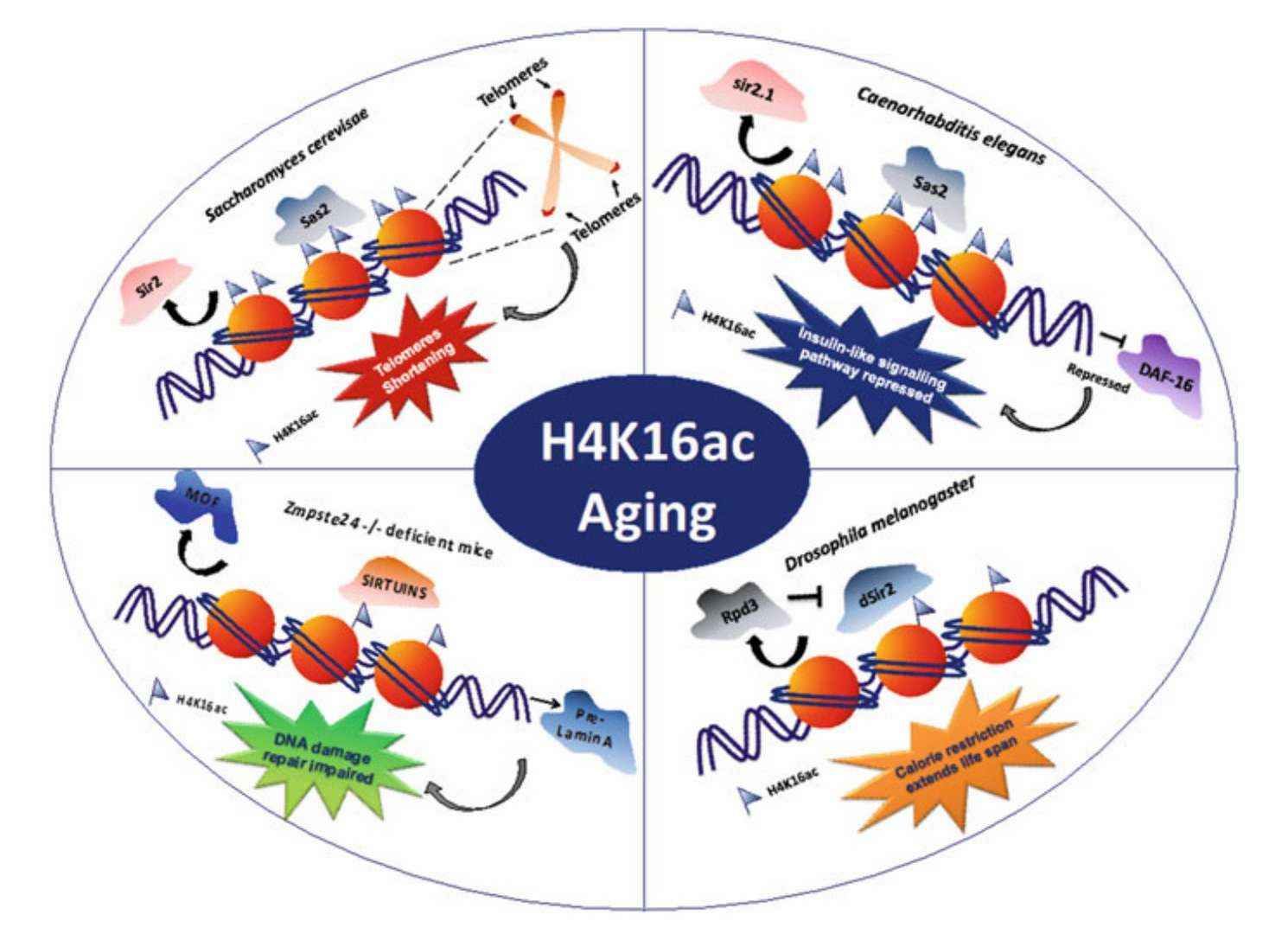 H4K16 acetylation in aging (Pandita et al., 2022)
H4K16 acetylation in aging (Pandita et al., 2022)
Synergistic effects of acetylation and chromatin remodeling
The synergistic interaction of SWI/SNF complexes (e.g., BRG1/BRM) with histone acetylation can be explained by a "two-step model." First, H3K27ac recruits SWI/SNF to the target gene region via Brd4. Second, SWI/SNF uses the energy generated by the hydrolysis of ATP (~50 kJ/mol) to This process involves the movement of nucleosomes, with an average of 10 base pairs (bp) per molecule of ATP hydrolyzed. This energetic coupling mechanism plays a critical role in various stages of embryonic development. For instance, in the zebrafish model, double knockdown of BRG1 and P300 resulted in complete heart arrest, whereas knockdown of either gene alone resulted in a mild phenotype, demonstrating that these two genes are functionally irreplaceable.
The dynamics and cellular heterogeneity of histone modifications have long posed significant challenges in the field of epigenetic research. Conventional techniques, such as chromatin immunoprecipitation sequencing (ChIP-seq), have yielded global maps of histone modifications; however, their resolution is constrained to a few hundred base pairs and necessitates millions of cell samples, impeding the resolution of heterogeneity at the single-cell level and the tracking of dynamic modifications. However, recent advancements in single-cell epigenomics and spatial technologies have emerged as game-changers in this field. These innovations have facilitated the revelation of regulatory networks of histone modifications in higher spatial and temporal dimensions, offering novel insights into disease mechanisms and precision therapies.
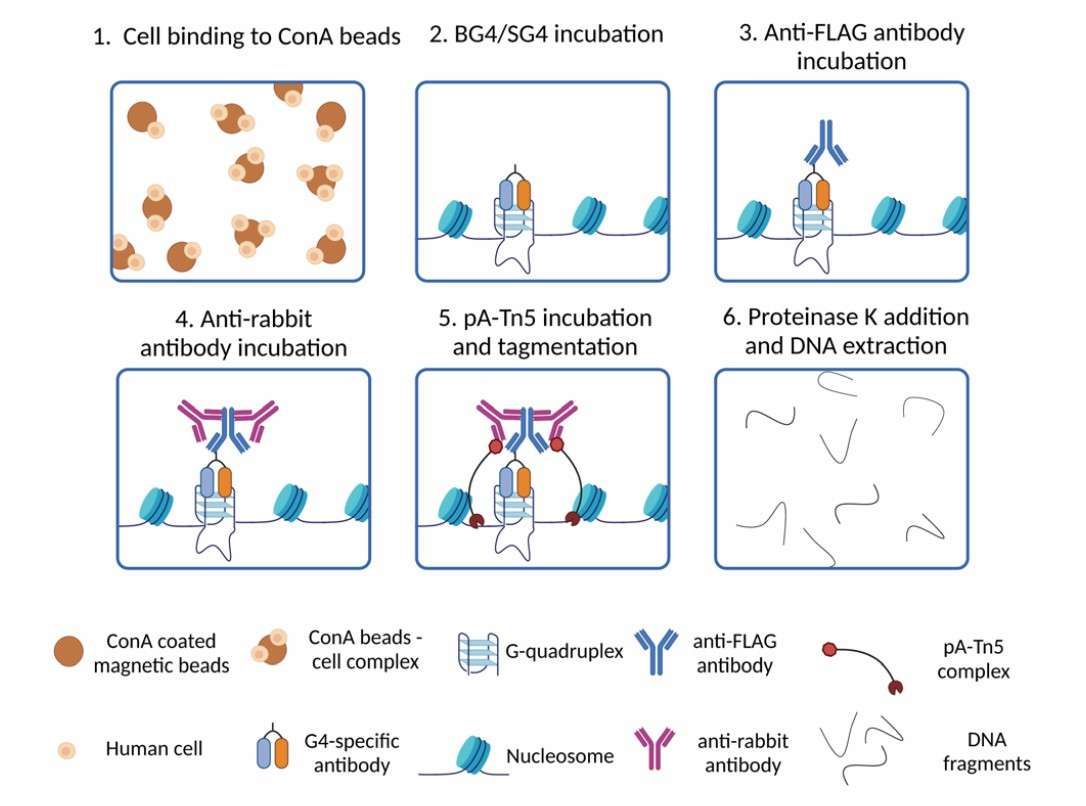 Schematic of G4 CUT&Tag workflow (Galli et al., 2024)
Schematic of G4 CUT&Tag workflow (Galli et al., 2024)
The CUT&Tag technology is a method that utilizes antibody-mediated targeted binding in combination with efficient cleavage by Tn5 transposase to achieve high-resolution detection of histone modifications. The underlying principle is to employ a specific antibody to recognize the target modification site (e.g., H3K4me3), which is then bound to an antibody through Protein A/G to guide the Tn5 transposase carrying the sequencing junction to precisely cut the target chromatin region. In comparison to conventional ChIP-seq, CUT&Tag exhibits not only an enhancement in resolution, from 500 bp to 50 bp, but also a substantial reduction in sample requirements, with only a few hundred cells being sufficient for completion of the experiment. This advancement facilitates epigenetic analysis of samples that are otherwise scarce, such as puncture biopsies or circulating tumor cells. A notable illustration of this advance is its application in lung cancer studies, where CUT&Tag successfully identified the aberrant enrichment of WNT pathway enhancer regions H3K4me3 and H3K27ac in EGFR-mutant tumor cells. This finding provides a theoretical foundation for combination therapies that target epigenetic drugs and kinase inhibitors.
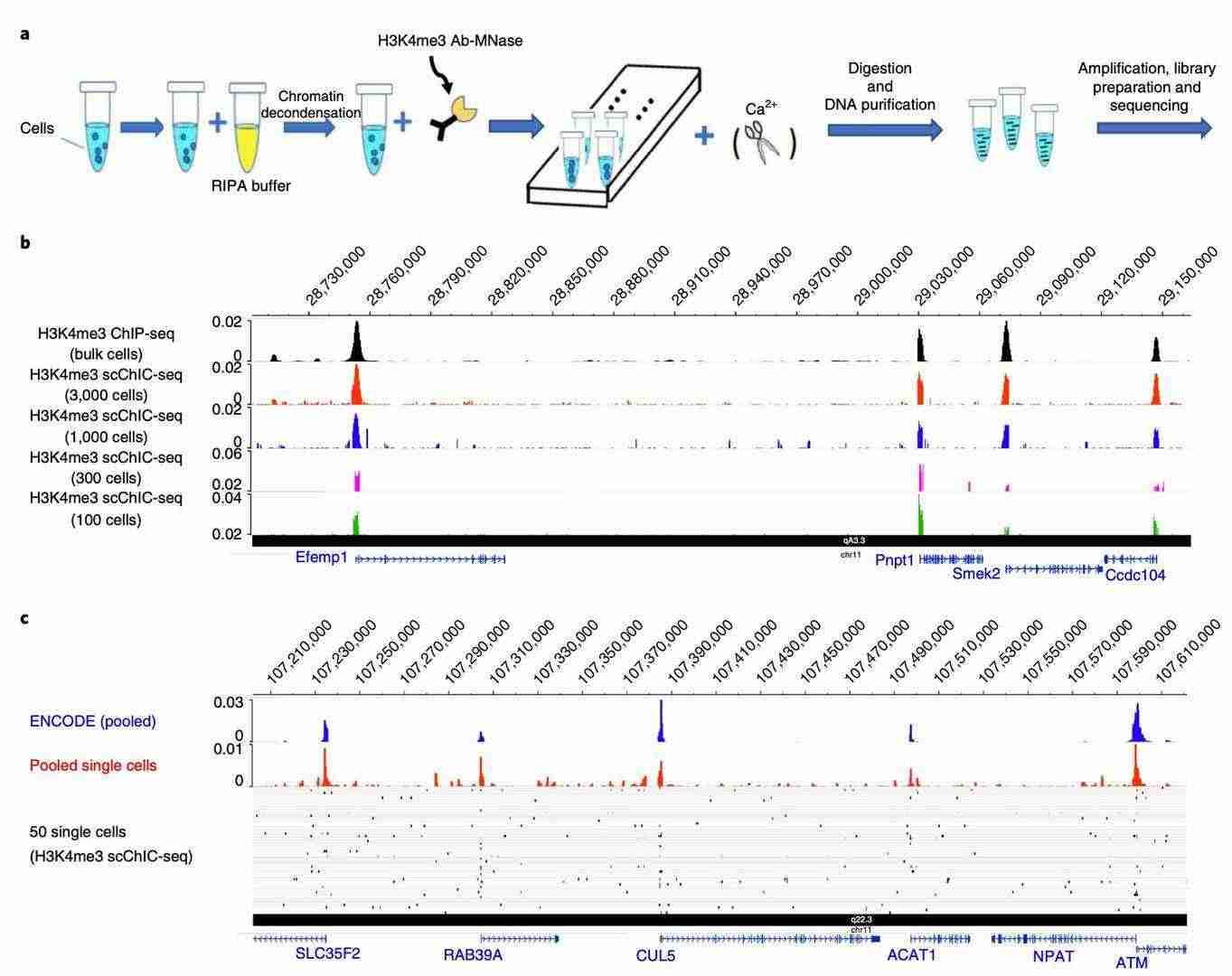 scChIC-seq detects H3K4me3 profiles in a small number of cells and single cells (Ku et al., 2019)
scChIC-seq detects H3K4me3 profiles in a small number of cells and single cells (Ku et al., 2019)
However, the challenge of single-cell heterogeneity remains unresolved. In addressing this need, single-cell histone modification sequencing technologies (e.g., scChIC-seq) have emerged. This technology utilizes microfluidic chips to encapsulate individual cells in water-in-oil microdroplets, labels specific modifications (e.g., H3K27me3) using antibodies carrying unique barcodes, and subsequently cleaves the chromatin and constructs sequencing libraries by MNase. This strategy not only resolves the apparent state of different cellular subpopulations in the tumor microenvironment, but also integrates single-cell transcriptomic data to reveal the dynamic association between modifications and gene expression. In the context of glioblastoma, scChIC-seq has led to the identification of three epigenetic subpopulations: quiescent stem cells, which are characterized by high levels of H3K27me3 and drug resistance; proliferating cells, which are marked by H3K4me3 and elevated PD-L1 expression; and invasive front cells, which are distinguished by H3K9ac enrichment and a link to angiogenesis. These findings suggest that combination therapies targeting epigenetic heterogeneity, such as H3K27me3 inhibitors in combination with immune checkpoint blockers, may be more effective in suppressing tumor recurrence.
Technological innovations have also driven the development of spatial epigenomics.10x Genomics Visium HD technology enables spatial localization at 4 μm resolution by combining tissue sections with histone-modifying antibodies. A notable finding from a study on breast cancer tissue sections revealed a significant deletion of H3K9ac in the core region of the tumor and an aberrant activation of H3K4me3 in the marginally invasive region. This finding suggests a spatial distribution difference that may be correlated with the tumor's propensity for immune escape and metastasis. However, it is imperative to acknowledge the limitations of the current techniques. The antibody cross-reactivity rate (~8%) may lead to the production of false-positive signals, while live cell dynamic tracking techniques (e.g., CRISPR-dCas9-based fluorescent reporter systems), despite their capacity to monitor changes in H3K9ac in real time, are constrained by fluorescence quenching and background noise.
In the future, the maturation of multi-omics integration and automation technologies will further enhance the depth of epigenetic research. For example, by combining single-cell epigenetic profiling, transcriptomic, and proteomic data, a three-dimensional regulatory network of "modification-expression-function" can be constructed, and artificial intelligence-driven epigenetic characterization is expected to mine new biomarkers from massive data. The clinical translational potential of these technologies has already been demonstrated in liquid biopsies, where the level of H3K27ac in circulating tumor cells can be used to predict a patient's therapeutic response to HDAC inhibitors. As epigenetics moves from basic research to precision medicine, technological innovations will continue to play a bridging role, connecting molecular mechanisms to clinical practice, and ultimately achieving the ultimate goal of individualized therapy.
Service you may intersted in
Learn More:
The development of epimedicines is confronted by two significant challenges: functional redundancy of modified enzymes and off-target effects. However, a novel generation of selective inhibitors and combination therapies is progressively overcoming these limitations.
HDAC inhibitors: remodeling the tumor immune microenvironment
The immunomodulatory mechanism of the HDAC inhibitor vorinostat involves multiple pathways, including: (1) enhancement of type I interferon signaling through acetylation of STAT1 to promote antigen presentation; (2) inhibition of Foxp3 expression in Treg cells to deregulate immunosuppression; and (3) induced up-regulation of MHC-I-like molecules in tumor cells. In the melanoma clinical trial, vorinostat in combination with pembrolizumab increased the objective remission rate (ORR) from 45% to 68%, and the treatment response was positively correlated with intra-tumor H3K27ac levels (r=0.72, p<0.01).
BET inhibitors: reversing oncogenic transcriptional programs
Research findings on the resistance mechanism of the BET inhibitor JQ1 revealed that leukemia cells can upregulate Brd4 protein expression (~3-fold) by activating non-classical NF-κB signaling, leading to a compensatory elevation of H3K27ac in the super-enhancer region. To address this phenomenon, researchers developed an "epigenetic synthetic lethality" strategy. This strategy entailed the combination of JQ1 and a CDK9 inhibitor (e.g., AZD4573). This combination synchronously blocked transcriptional elongation and enhancer activity. This resulted in a 90% reduction of tumor load in a mouse model of AML. This regimen is currently undergoing phase I clinical trials, and preliminary data has shown 67% clearance of peripheral blood primitive cells.
Recent advancements in CUT&Tag and single-cell histone modification sequencing technologies have facilitated a paradigm shift in research methodologies, enabling researchers to transition from the conventional "population averaging" approach to a more precise "single-cell precision" analysis. This transition, coupled with the ability to generate "dynamic movies" rather than the traditional "static snapshots," signifies a significant evolution in biological research. These technologies are not only propelling fundamental research, such as elucidating enhancer-promoter interactions, but also exemplifying their potential for clinical translation. A notable illustration of this is the use of liquid biopsy to detect the epigenetic features of circulating tumor cells, facilitating early screening and efficacy prediction. As automation and multi-omics integration continue to evolve, epigenetics research stands on the precipice of a new era, one characterized by precision medicine.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.